© Л.Л.Кіселев
ЯК ЗУПИНИТИ СИНТЕЗ БІЛКІВ?
Л.Л. Кисельов
Лев Львович Кисельов, академік РАН і Європейської академії (Academia Europaea),
завідувач лабораторією Інституту молекулярної біології ім.В.А.Енгельгардта РАН.
Завдяки досягненням молекулярної біології останніх двох десятиліть ми знаємо, як ініціюється білковий синтез на рибосомах (клітинних частинках, в яких протікає трансляція, тобто синтез білків) і як відбувається подовження поліпептидного ланцюга (елонгація). Однак завершення синтезу (термінація) залишалося поза увагою дослідників, хоча ще з кінця 1960-х років відомі два основних факту. По-перше, синтез припиняється, коли в ділянці А малої субодиниці рибосоми з'являється стоп (нонсенс-) кодон. Це один з трьох трінуклеотідамі (УАА, УАГ, УГА), які не кодують амінокислот, а служать сигналом для закінчення елонгації (рис.1). По-друге, "зчитування" стоп-кодонів в рибосомі потребує особливих білках, названих факторами звільнення поліпептидних ланцюгів (release factors, скорочено RF). За властивостями і ролі в клітці ці білки поділяються на два класи. У бактерій відомі два фактора термінації класу-1 - RF1 (довідається стоп-кодони УАА та УАГ) і RF2 (довідається УАА та УГА), у архей - aRF1, у евкаріот - eRF1 (узнающие все три стоп-кодону).
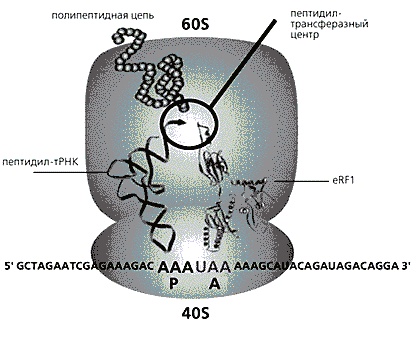
Мал. 1. Схема термінації білкового синтезу у евкаріот.
У міру того, як молекула мРНК просувається крізь рибосому (що складається з 60S і 40S субчастиц), її нуклеотидних послідовність перекладається в амінокислотну. Присутність стоп-кодону UAA і eRF1 в А-ділянці рибосоми призводить до зупинки синтезу - розщепленню пептидил-тРНК (звільнення поліпептидного ланцюга). До кінця 80-х років амінокислотна послідовність багатьох бактеріальних факторів стала відома, а от з eRF1 сталася драматична історія, про яку я вже розповідав [ 1 ]. Американські дослідники в результаті методичних помилок приписали йому структуру зовсім іншого білка. Виявивши це [ 2 ], Ми змогли розшифрувати справжню послідовність амінокислот eRF1 [ 3 ], Що дозволило ідентифікувати схожі на нього архебактеріальной фактори aRF1. При цьому з'ясувалося, що eRF1 різко відрізняється по структурі від раніше відомих RF1 і RF2, і ми припустили, що фактори термінації бактерій і евкаріот походять від різних предкових молекул [ 3 ]. Така гіпотеза викликала різку критику з боку деяких дослідників. Однак подальше вивчення великого числа структур [ 4 ], А також рентгеноструктурний аналіз кристалів eRF1 (виконаний в Англії) і RF2 (в Данії) повністю підтвердили нашу правоту.
Структуру факторів класу-2 бактеріального RF3 [ 5 ] І евкаріотіческого eRF3 [ 6 ] Встановили майже одночасно. Вони виявилися ферментами, які каталізують всередині рибосом гідроліз гуанозінтріфосфата (ГТФ), не беруть участі в термінації (розщепленні кінцевого продукту білкового синтезу, пептидил-тРНК), але сприяють звільненню з рибосом факторів класу-1 [ 7 , 8 ].
Ключова роль трипептида GGQ
Бактеріальні та евкаріотіческіе фактори класу-1, незважаючи на несхожість своїх первинних структур, мають один спільний елемент - трипептид гліцил-гліцил-глютамин (скорочено GGQ). Він присутній у всіх білках класу-1, а їх зараз відомо для бактерій, архей і евкаріот вже близько 100 (рис.2). Виявивши спільно з колегами з центру по вірусології та біотехнології "Вектор" цей універсальний мотив, ми припустили, що він відповідальний за хімічну стадію термінації (розщеплення пептидил-тРНК) [ 9 ]. Це означало, що його зміна призведе до втрати активності фактора, але не завадить йому зв'язуватися з рибосомою, пошкоджений білок перетвориться в інгібітор термінації. Сам трипептид GGQ повинен перебувати у великій субчастиц рибосоми, в області її пептидилтрансферазного центру. При експериментальній перевірці всі ці слідства повністю підтвердилися.
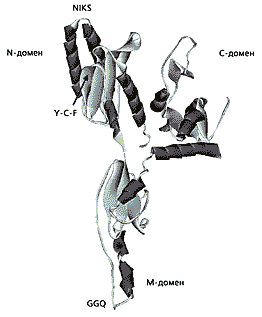 Мал.2. Схема тривимірної структури eRF1 людини
Мал.2. Схема тривимірної структури eRF1 людини
за даними рентгеноструктурного аналізу [ 12 ].
N-домен дізнається стоп-кодон, M-домен знаходиться в пептидилтрансферазної центрі рибосом і бере участь в гідролізі пептидил-тРНК, С-домен зв'язується з фактором класу-2 eRF3. Трипептид GGQ знаходиться на одному з кінців молекули eRF1, утворюючи гнучку петлю. Таке просторове розташування забезпечує контакт eRF1 з пептидил-тРНК, яка розташовується в сусідньому з молекулою ділянці (Р).
Таким чином, вдалося показати, що GGQ служить активним центром чинників класу-1 (у прокаріот і евкаріот) і, розташовуючись в пептидилтрансферазної центрі, каталізує розщеплення ефірного зв'язку в пептидил-тРНК. Цікаво, що ключова роль цього трипептида в термінації забезпечується в першу чергу двома гліциновими залишками, а не глютамином [ 10 - 15 ]. Можливо, що відсутність у гліцину бічний групи звільняє простір для молекули води, що бере участь в гідролізі пептидил-тРНК, оскільки заміни гліцинового залишків на більш об'ємні незмінно ведуть до втрати активності у мутантних RF.
Як чинники дізнаються стоп-кодони?
Фактор класу-1 каталізує гідроліз, тільки якщо в А-ділянці малої субодиниці присутній один з трьох стоп-кодонів. Іншими словами, всередині рибосоми сигнал від стоп-кодону (що знаходиться в малій субчастиц) до пептидилтрансферазної центру (розташованому в великий рибосомной субчастиц) передається через GGQ-мотив. Як така передача може здійснюватися? Згідно з однією гіпотезою, стоп-кодон специфічно зв'язується з ділянкою малої Хвороби, а фактор відіграє лише допоміжну роль. Відповідно до іншої, стоп-кодон взаємодіє з "який дізнається" трипептидом фактора класу-1. У першому випадку головну роль в передачі сигналу грає рибосома, а в другому - фактор класу-1. На підставі спільних дослідів з французькими [ 16 ] І японськими [ 17 ] Дослідниками ми віддали перевагу другою гіпотезою. Виявилося: якщо в пробірці створити "гібрид" з рибосоми, яка вміє "зчитувати" все три стоп-кодону, і фактора класу-1, виділеного з організму, де використовуються тільки один або два стоп-кодону, то результат буде залежати від природи чинника. Звідси можна припускати прямий контакт eRF1 зі стоп-кодоном в рибосомі. Дійсно, використовуючи синтезовані триплети з хімічно модифікованими групами, ми спільно з паризькими і новосибірськими колегами довели, що вони утворюють ковалентний зв'язок з молекулою eRF1 всередині рибосоми [ 18 , 19 ]. Це можливо лише в тому випадку, якщо вони зближені всередині рибосоми. Аналогічні результати були отримані іншими дослідниками для факторів класу-1 і рибосом бактерій. Отже, чинники класу-1 повинні мати ділянку, специфічно що довідався в рибосомі стоп-кодони мРНК.
Нещодавно таку ділянку вдалося знайти у RF1 і RF2 бактерій [ 20 ]. Це були трипептиди, але різні (оскільки RF1 дізнається УАГ, а RF2 - УГА, а УАА дізнаються обидва чинники). Поки не показано, чи знаходяться вони в безпосередньому контакті зі стоп-кодоном в рибосомі. Однак результат нічого не дав для розуміння термінації у евкаріот, оскільки первинні і просторові структури цих факторів класу-1 у двох царств живої природи влаштовані по-різному.
Виходячи з того, що відстань між стоп-кодоном і пептидилтрансферазної центром рибосом становить приблизно 75 Е (це випливає з форми і розмірів тРНК і даних рентгеноструктурного аналізу рибосом прокариот), що довідався ділянку може розташовуватися або на N-кінцевому, або на С-кінцевому доменах . Друге припущення відпадає відразу ж, оскільки видалення С-домена не заважає пізнанню: такий укорочений білок в пробірці в повній мірі зберігає свою активність [ 21 ]. Значить, стоп-кодон повинен дізнаватися N-доменом, що підтверджується дослідами з мутантним [ 22 , 23 ] І "гібридним" eRF1 [ 17 ], А також даними інших дослідників.
Однак поки не ясно, де саме знаходиться ця ділянка, так як розмір N-домену досить великий. У пошуках відповіді ми проаналізували амінокислотну послідовність факторів eRF1 з самих різних організмів (за даними з загальнодоступних банків). Два пептидних фрагмента, АСН-Ілі ---- Ліз-Сер (NIKS) і Тир-Цис --- Фен (YCF) *, розташовані в петлях N-домену (повністю або частково), що полегшує їм контакт зі стоп-кодоном в рибосомі. Присутність в пептидах консервативних і напівконсервативним амінокислотних залишків теж суперечить гіпотезі про їх можливу роль як дізнаються стоп-кодон. Однак потрібні були прямі експериментальні докази, які ми отримали за допомогою спрямованого мутагенезу.
* Серія тире означає, що між цими інваріантними амінокислотами знаходяться інші, які варіюють. Ми замінили в тетрапептид NIKS кожну амінокислоту і подивилися, як це позначається на функціональній активності eRF1 людини. Досліди, поставлені в пробірці, дали цілком чіткі результати [ 22 ]. Заміна изолейцина (I) досить серйозно зменшує активність мутантного білка; ефект від заміни лізину (К) набагато м'якше; крайні амінокислоти аспарагін (N) і серин (S) займають проміжні положення. В цілому, фрагмент NIKS безумовно функціонально важливий, але стверджувати на підставі наших даних, що саме він дізнається весь стоп-кодон, навряд чи правильно.
Зовсім іншу картину ми отримали, коли аналізували ділянку YCF [ 23 ]. При заміні цистеїну (С) або фенілаланіну (F) на інші амінокислоти відповідь на стоп-кодон УГА майже не змінювався, а на УАА та УАГ падав дуже сильно. Більш того, якщо до кожної з цих мутацій додати ще по одній - по аспарагін (N), що стоїть між C і F, то eRF1 людини стає схожим на eRF1 реснитчатих найпростіших. У них фактор дізнається тільки УГА, а два інших триплета (УАА та УАГ) перетворюються в значущі, що кодують амінокислоти. Такі білки, які дізналися тільки один стоп-кодон, називають уніпотентнимі, на відміну від омніпотентних eRF1 вищих евкаріот.
Отже, спрямована зміна структури eRF1 в області YCF-петлі призвело до втрати дізнатися здатності фактора щодо двох стоп-кодонів, зберігши її в відношенні третього, УГА. Значить, і цистеїнових, і фенілаланіновой залишки, безумовно, беруть участь в впізнаванні стоп-кодонів, хоча поки не відомо, прямо або побічно. Оскільки у двох триплетів, УАА та УАГ, на відміну від УГА, загальний елемент - другий нуклеотид (аденін), цілком можливо, що саме в його впізнавання беруть участь цистеїн і фенілаланін.
Однак такий висновок суперечить тому відомому обставині, що в організмах з варіантних генетичними кодами (реснитчатих найпростіших) впізнається тільки частина стоп-кодонів, а ці амінокислоти проте присутні в структурі їх чинників. Щоб пояснити таку ситуацію, ми запропонували концепцію негативних елементів, які перешкоджають у реснитчатих участі цистеїну і / або фенілаланіну в впізнаванні стоп-кодонів (можливо, аденіну у другій позиції). Дійсно, якщо подивитися на амінокислоти, що оточують цистеїн і фенілаланін зліва і справа, видно (рис.3), що вони у організмів з універсальним генетичним кодом (вищих евкаріот) відрізняються від таких з варіантних кодами. Деякі з цих амінокислот могли б виконувати роль таких негативних елементів. Зрозуміло, наше припущення вимагає експериментальної перевірки. Однак щось подібне досить добре вивчено на прикладі тРНК і аміноацил-тРНК-синтетаз, де в тРНК окремі нуклеотиди якраз і служать негативними елементами.

Перетворення омніпотентного фактора (eRF1 людини) в уніпотентний (характерний для деяких найпростіших евкаріот, що довідався лише УГА), здійснене за допомогою точкового мутагенезу, швидше за все імітує процес, що відбувався в живій природі десятки мільйонів років тому. Згідно поглядам еволюціоністів, організми з варіантних кодами походять від організмів з універсальним (стандартним) генетичним кодом, а не навпаки. Іншими словами, раніше фактори термінації реснитчатих вміли дізнаватися все стоп-кодони, а потім "розучилися". Чи можна будь-яким способом переконатися, що такий потенціал у них зберігся? Виявилося, що універсальність впізнавання можна повернути, причому вкрай просто - змінивши температуру культивування організму [ 17 ]. Цей досвід разом з іншими підтверджує два припущення: по-перше, між факторами термінації з організмів з універсальним і варіантних кодами немає прірви (при деяких обставинах вони взаємно перетворюваність). По-друге, якщо нейтралізувати дію негативних елементів (в даному випадку зміною температури), впізнаються все стоп-кодони.
Відповідно до гіпотези білкового антикодону, запропонованої для прокаріотів, трипептиди PAT (Про-Ала-Тре) в RF1 або SPF (Сер-Про-Фен) в RF2 дізнаються по два стоп-кодону з трьох [ 20 ]. Чи можна застосувати концепція білкового антикодону (тобто короткою лінійної послідовності амінокислот, дізнавшись трінуклеотідамі) до eRF1?
Наші досліди швидше говорять про зворотне [ 23 ]. Перетворення омніпотентного фактора в уніпотентний вдалося досягти, не тільки змінюючи цистеїн або фенілаланін, а й інші амінокислоти в двох мінідоменах NIKS і YCF одночасно. Зміни окремо в кожному з них є недостатніми для подібного ефекту (рис.4). Значить, два мінідомена, рознесених в просторі, при впізнаванні стоп-кодонів взаємодіють один з одним. Це вже не лінійна безперервна послідовність амінокислот, як передбачається для RF1 і RF2. Мабуть, крім глибоких відмінностей в структурі фактори класу-1 прокаріотів і евкаріот розрізняються і за механізмом пізнавання.

новий метод
Вивчення молекулярного механізму термінації потребує адекватних експериментальних підходах. Методи, якими користуються багато дослідників, для цих цілей вже недостатньо досконалі. Сьогодні необхідний метод, який об'єднував би умови досвіду in vivo, існуючі в клітці, і in vitro - в пробірці, коли експериментатор може змінювати умови досвіду за своїм бажанням. Вирішуючи таку непросту задачу [ 24 ], Ми спиралися на показану нами разом з французькими дослідниками in vitro конкуренцію за стоп-кодони між eRF1 і супресорних тРНК [ 25 ], Які можуть "читати" стоп-кодони як значущі (кодують ту чи іншу амінокислоту). Цей принцип дозволяє оцінювати кількісно властивості як eRF1 (і, зрозуміло, його мутантів), так і супресорних тРНК. В якості матриці, яка була пов'язана з рибосомами, ми використовували копію гена люциферази (ферменту, вплив якого на субстрат призводить до випускання кванта світла). Якщо матриця "зчитується" повністю, утворюється активний білок, який змушує люциферин (субстрат) добре світитися в рибосомной системі, забезпеченою всіма необхідними компонентами. Якщо ж в матрицю вставити стоп-кодон, синтез поліпептиду обривається, і світіння субстрату немає. Додаючи в таку систему eRF1, супресорні тРНК і їх суміші, можна "на замовлення" міняти інтенсивність синтезу люціферази і за світінням вимірювати її активність. Ця ж система годиться і для вивчення властивостей і активності eRF3.
Тепер ми маємо такий метод оцінки властивостей eRF1, який в подальшому дозволить по-новому міряти кінетику термінації і здійснювати варіанти аналізу, нереальні при використанні традиційних підходів.
Можливості молекулярної терапії
Феномен конкуренції eRF1 і супресорних тРНК [ 24 , 25 ] Наштовхнув нас ще на одну ідею, що стосується біомедицину або, як зараз модно говорити, до молекулярної медицини. Багато спадкові хвороби пов'язані з тим, що в результаті пошкодження гена один з значущих триплетів перетворюється в незначний (такі мутації називають нонсенс-мутаціями). Цей стоп-кодон, потрапляючи в мРНК, призводить до синтезу укороченого білка, який, як правило, біологічно неактивний.
Теоретично існують два принципово різних шляхи усунення такого дефекту. Перший - генотерапія, тобто заміщення пошкодженого гена нормальним, що не містить в кодує області стоп-кодону. Методи генотерапіі зараз активно розробляються, але, на жаль, до сих пір без особливого успіху. Другий шлях пов'язаний з виправленням генного продукту - білка.
Як можна вілікуваті хворий білок? Очевидно, нужно НЕ дати спрацюваті стоп-кодону Всередині мРНК (тобто Забезпечити ДОБУДОВУ поліпептідного ланцюга до кінця), что можливо двома способами. Перший - блокуваті будь-Якім чином eRF1 (например, додали до него антитіла або відповідній інгібітор). Терминация буде ослаблена, и в дію вступлять супресорні тРНК, Які в невеликих кількостях всегда Присутні в клітінах Вищих організмів. Другий - внести в систему супресорні тРНК, які за рахунок конкуренції витіснять eRF1 з А-ділянки рибосоми, а стоп-кодон буде прочитаний як значущий. Це, швидше за все, призведе до синтезу повнорозмірного білка з нормальною активністю.
Обидва підходи реалізовані нами експериментально. За допомогою популярного методу Selex ми спільно з вченими США отримали набір коротких молекул РНК (Аптамерів), які блокують активність фактора eRF1 in vitro [ 26 ]. Є надія, що якщо такі інгібітори-аптамери ввести в клітини хворого, термінація буде ослаблена і хворий білок вилікується.
Спільно з Інститутом акушерства і гінекології ім.Д.О.Отта РАМН в Санкт-Петербурзі [ 27 ] На мишах, у яких пошкоджений ген, що кодує білок дистрофин, ми показали, що після введення гена супрессорной тРНК в м'язові клітини в них з'являється дистрофин. Іншими словами, коли в клітинах зростає концентрація супрессорной тРНК, eRF1 витісняється з А-ділянки рибосоми і частина поліпептидних ланцюгів зчитується до кінця, без обриву на місці стоп-кодону.
Таким чином, способи впливу на терминацию в принципі можуть стати основою молекулярної терапії всіх спадкових і неспадкових дефектів білків, пов'язаних з передчасним завершенням синтезу. Звичайно, доведеться подолати ще багато труднощів на цьому шляху, і до лікування хворих поки далеко. Однак вперше в історії медицини з'явилися, по крайней мере ідеї про те, як можна підступитися до хвороб, які досі вважалися невиліковними.
Крім своєї основної функції - участі в передачі сигналу від стоп-кодону до пептидилтрансферазної центру рибосоми eRF1 грають ще одну важливу роль: вони активують ГТФазную активність фактора eRF3 класу-2. Фактори eRF1 і eRF3 людини взаємодіють один з одним своїми С-кінцевими ділянками, утворюючи досить міцні комплекси, вони виявлені як in vitro, так і in vivo, причому незалежно від присутності рибосом і стоп-кодонів [ 21 , 28 , 29 ]. У евкаріот eRF3 каталізує розщеплення ГТФ тільки при одночасному присутності рибосом і eRF1 [ 7 ]. Така ж ГТФазная активність відзначена і для бактеріальних факторів RF3. Однак якщо eRF1 і eRF3 in vitro дають досить міцний комплекс, то у прокаріотів такого комплексу поки ніхто не отримав.
Після виявлення цього феномена з'явилися дві гіпотези [ 30 ] Про можливу роль факторів класу-1 щодо факторів класу-2. Згідно з першою, вона полягає в активації ферменту шляхом "добудови" його активного центру. Друга гіпотеза постулює, що фактори класу-1, зв'язуючись з ГТФазамі, різко прискорюють обмін ГТФ-ГДФ на ферменті, що збільшує каталітичну активність. Перша гіпотеза ще потребує експериментальної перевірки [ 31 ], Друга - експериментально підтверджена для бактеріального eRF3 [ 32 ].
* * *
Підіб'ємо короткі підсумки 10-річного вивчення термінації білкового синтезу у евкаріот.
Розкрито структуру (первинна, вторинна, просторова) білка eRF1 - фактора термінації класу-1.
Встановлено, що він визначає специфічність зчитування стоп-кодонів, взаємодіючи своїм N-кінцевим доменом зі стоп-кодоном в рибосомі.
Виявлено амінокислоти, які є важливими для функцій eRF1 в рибосомі.
Доведено, що універсальний симетричний трипептид Гли - Гли-Глн грає вирішальну роль при розщепленні пептидил-тРНК. Феномен конкуренції між eRF1 і супресорних тРНК за стоп-кодон в А-ділянці рибосоми ліг в основу нового методу визначення активності фактора і нового підходу до лікування спадкових хвороб людини, пов'язаних з нонсенс-мутаціями.
Отримано виборчі інгібітори eRF1 (РНК-аптамери).
Встановлено первинна структура і біологічна функція фактора термінації класу-2 - eRF3.
Виявлено контакт між факторами двох класів eRF1 і eRF3 in vitro і in vivo і розкрито механізм їх взаємодії через С-кінцеві домени цих білків.
Сформульовано гіпотезу про біологічну роль eRF3 в клітинах.
На молекулярному рівні визначені відмінності між термінації білкового синтезу у прокаріотів і евкаріот.
Досягнення російської фізико-хімічної біології в цій області повністю визнані світовою науковою спільнотою, що легко простежити за недавніми оглядам цієї проблеми [ 30 , 31 , 33 - 36 ].
література
1. Кисельов Л.Л. Детективна історія з життя молекулярних біологів // Природа. 1993.? 7. С.91-95.
2. Frolova L., Dalphin M., Justesen J. et al. // EMBO J. 1993. V.12. P.4013-4019.
3. Frolova L., Goff X. le., Rasmussen HH et al. // Nature. 1994. V.372. P.701-703.
4. Кисельов Л.Л., Опаріна Н.Ю., Фролова Л.Ю. // молекулярну. біологія. 2000. Т.34. С.427-441.
5. Grentzmann G., Brechemier-Baey D., Heurgue V. et al. // Proc. Natl. Acad. Sci. USA. 1994. V.91. P.5848-5852.
6. Zhouravleva G., Frolova L., Goff X.le et al. // EMBO J. 1995. V.14. P.4065-4072.
7. Frolova L., Goff X.le, Zhouravleva G. et al. // RNA. 1996. V.2. P.334-341.
8. Freistroffer DV, Pavlov MY, MacDougаll J. et al. // EMBO J. 1997. V.16. P.4126-4133.
9. Frolova LY, Tsivkovskii RY, Sivolobova GF et al. // RNA. 1999. V.5. P.1014-1020.
10. Сеит-Небі А., Фролова Л., Іванова Н. та ін. // молекулярну. біологія. 2000. Т.34. С.899-900.
11. Seit-Nebi A., Frolova L., Justesen J., Kisselev L. // Nucl. Acids Res. 2001. V.29. P.3982-3987.
12. Song H., Mugnier P., Das AK et al. // Cell. 2000. V.100. P.311-321.
13. Zavialov AV, Mora L., Buckingham RH et al. // Mol. Cell. 2002. V.10. ? 4. P.789-798.
14. Mora L., Heurgue-Hamard V., Champ S. et al. // Mol. Microbiol. 2003. V.47 (1). P.267-275.
15. Janzen DM, Frolova L., Geballe A. // Mol. Cell. Biol. 2002. V.22. ? 24. P.8562-8570.
16. Kervestin S., Frolova L., Kisselev L. et al. // EMBO Rep. 2001. V.2. P.680-684.
17. Ito K., Frolova L., Seit-Nebi A. et al. // Proc. Natl. Acad. Sci. USA. 2002. V.99. P.8494-8499.
18. Chavatte L., Frolova L., Kisselev L. et al. // Eur. J. Biochem. 2001. V.268. P.2896-2904.
19. Bulygin KN, Repkova MN, Ven'yaminova AG et al. // FEBS Letters. 2002. V.514. P.96-101.
20. Ito K., Uno M., Nakamura Y. // Nature. 2000. V.403. P.680-684.
21. Frolova LY, Merkulova TI, Kisselev LL // RNA. 2000. V.6. P.381-390.
22. Frolova L., Seit-Nebi A., Kisselev L. // RNA. 2002. V.8. P.129-136.
23. Seit-Nebi A., Frolova L., Kisselev L. // EMBO Rep. 2002. V.3. P.881-886.
24. Мазур А.М., Холод Н.С., Сеит-Небі А.С. та ін. // молекулярну. біологія. 2002. т.36. С.129-135.
25. Drugeon G., Jean-Jean O., Frolova L. et al. // Nucleic Acids Res. 1997. V.25. P.2254-2258.
26. Carnes J., Frolova L., Zinnen S. et al. // RNA. 2000. V.6. P.1468-1479.
27. Кисельов А.В., Остапенко О.В., Рогожкіна Є.В. та ін. // молекулярну. біологія. 2002. т.36. С.43-47.
28. Frolova L., Simonsen J., Merkulova T. et al. // Eur. J. Biochem. 1998. V.256. P.36-44.
29. Merkulova TI, Frolova LY, Lazar M. et al. // FEBS Lett. 1999. V.443. P.41-47.
30. Buckingham R., Grentzmann G., Kisselev L. // Mol. Microbiol. 1997. V.24. P.449-456.
31. Kisselev LL, Ehrenberg M., Frolova L.Yu. // EMBO J. 2003. V.22. ? 2. P.175-182.
32. Zavialov AV, Buckingham RH, Ehrenberg M. // Cell. 2001. V.107. P.115-124.
33. Kisselev LL, Buckingham RH // Trends Biochem. Sci. 2000. V.25. P.561-566.
34. Ehrenberg M., Tenson T. // Nature Struct. Biol. 2002. V.9. P.85-87.
35. Janzen DM, Geballe AP Cold Spring Harbor Symp. Quant. Biol. Cold Spring Harbor, 2001. P.459-467.
36. Kisselev LL Structure. 2002.
Як чинники дізнаються стоп-кодони?
Як така передача може здійснюватися?
Чи можна будь-яким способом переконатися, що такий потенціал у них зберігся?
Чи можна застосувати концепція білкового антикодону (тобто короткою лінійної послідовності амінокислот, дізнавшись трінуклеотідамі) до eRF1?
Як можна вілікуваті хворий білок?